1L PADCEV® + PEMBROLIZUMAB EFFICACY
1L PADCEV® + PEMBROLIZUMAB EFFICACY
A standard of care across 1L la/mUC1-4
EFFICACY
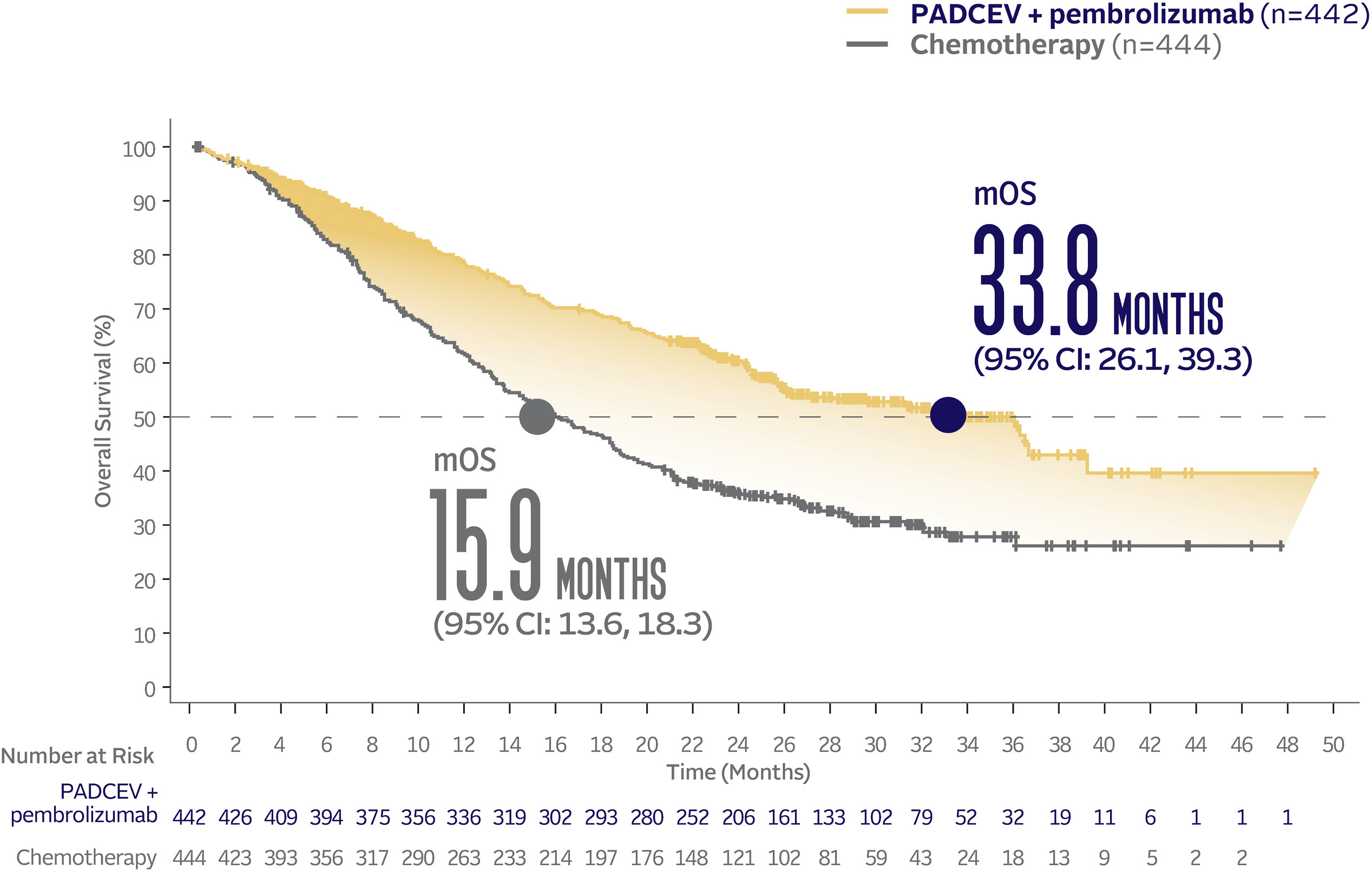
PADCEV + pembrolizumab nearly doubled mOS vs chemotherapy1
PADCEV + pembrolizumab reduced the risk of death by 53% vs chemotherapy (HR=0.47; 95% CI: 0.38, 0.58†; P<0.0001)‡
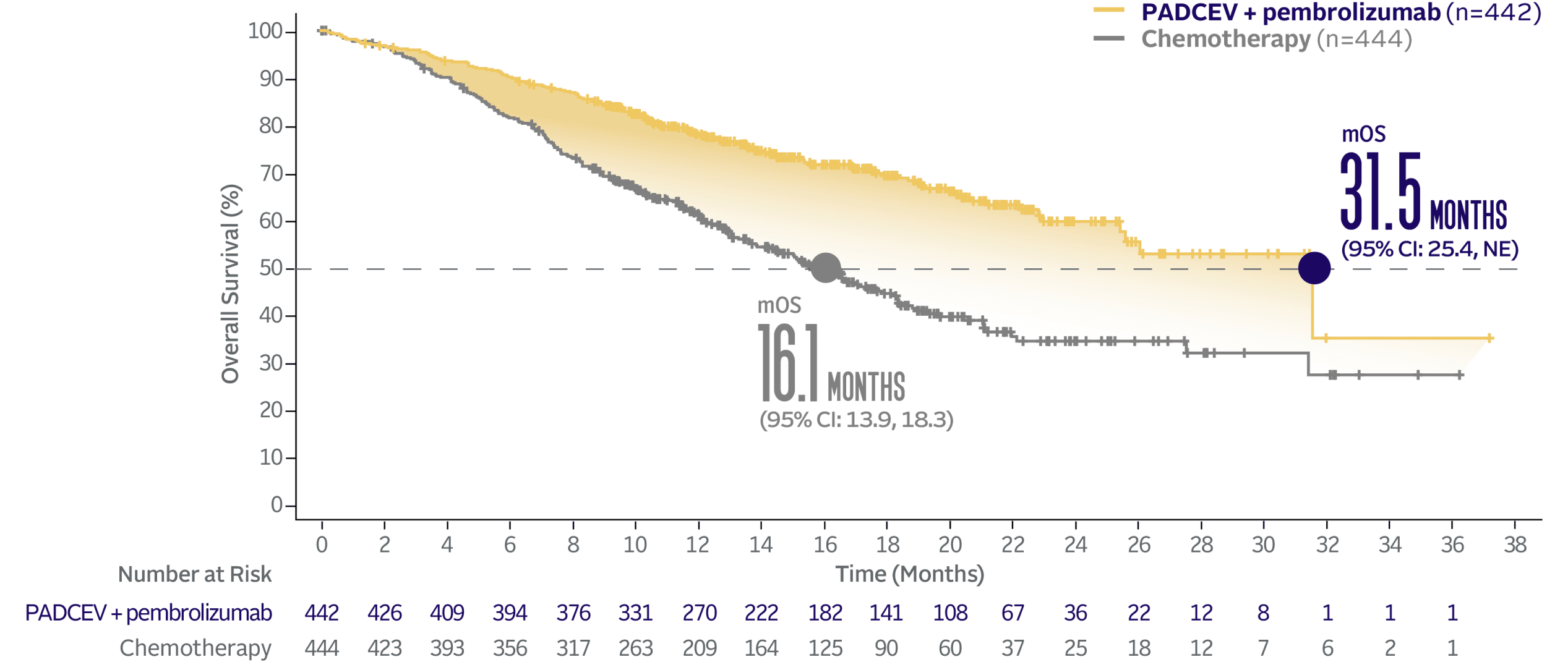
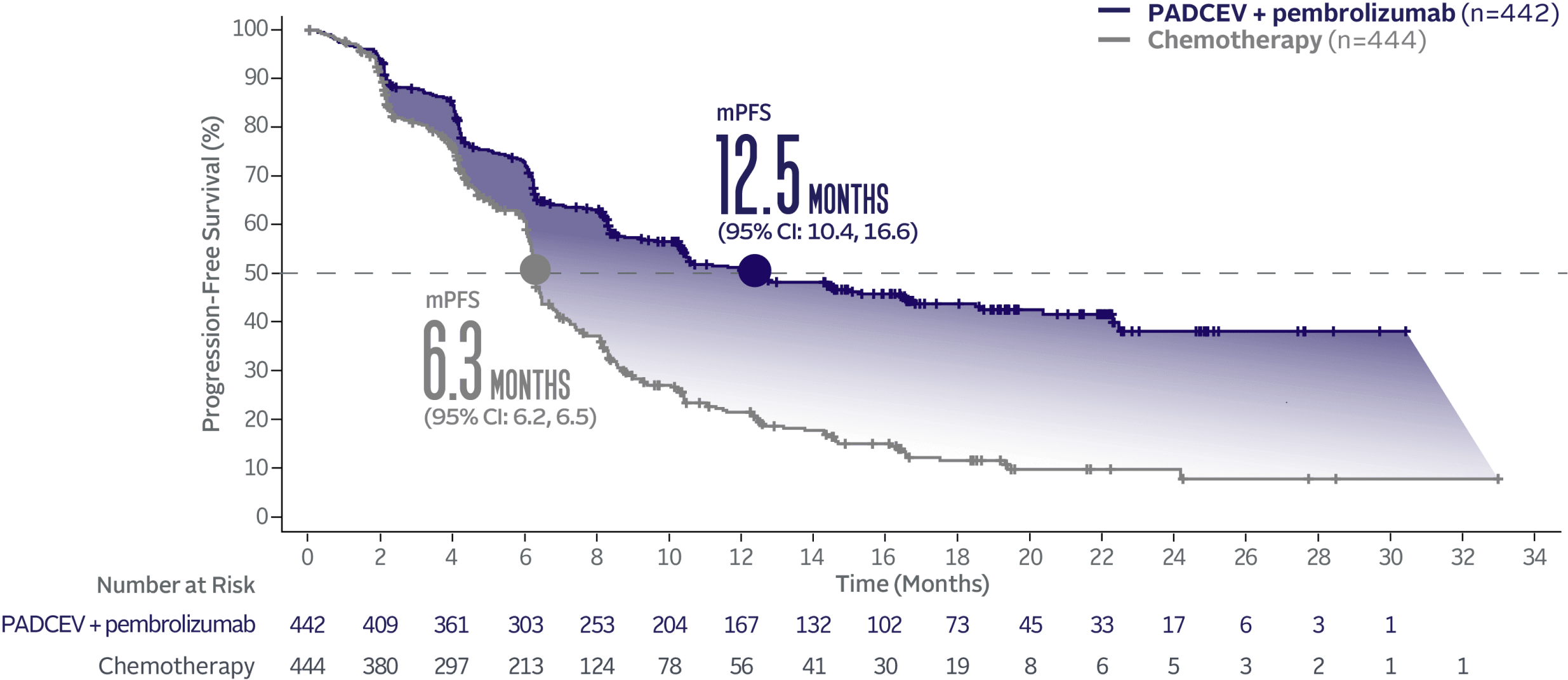
The median survival follow-up time was 17.2 months (range: 0.1, 37.2).2,5
Enfortumab vedotin-ejfv (PADCEV), in combination with pembrolizumab, is recommended by the National Comprehensive Cancer Network® (NCCN®) for adult patients with la/mUC6

The only NCCN Category 1 Preferred 1L treatment option
for la/mUC regardless of cisplatin eligibility
PADCEV + pembrolizumab nearly doubled mPFS vs chemotherapy1
PADCEV + pembrolizumab reduced the risk of progression or death by 55% vs chemotherapy (HR=0.45; 95% CI: 0.38, 0.54†; P<0.0001)‡
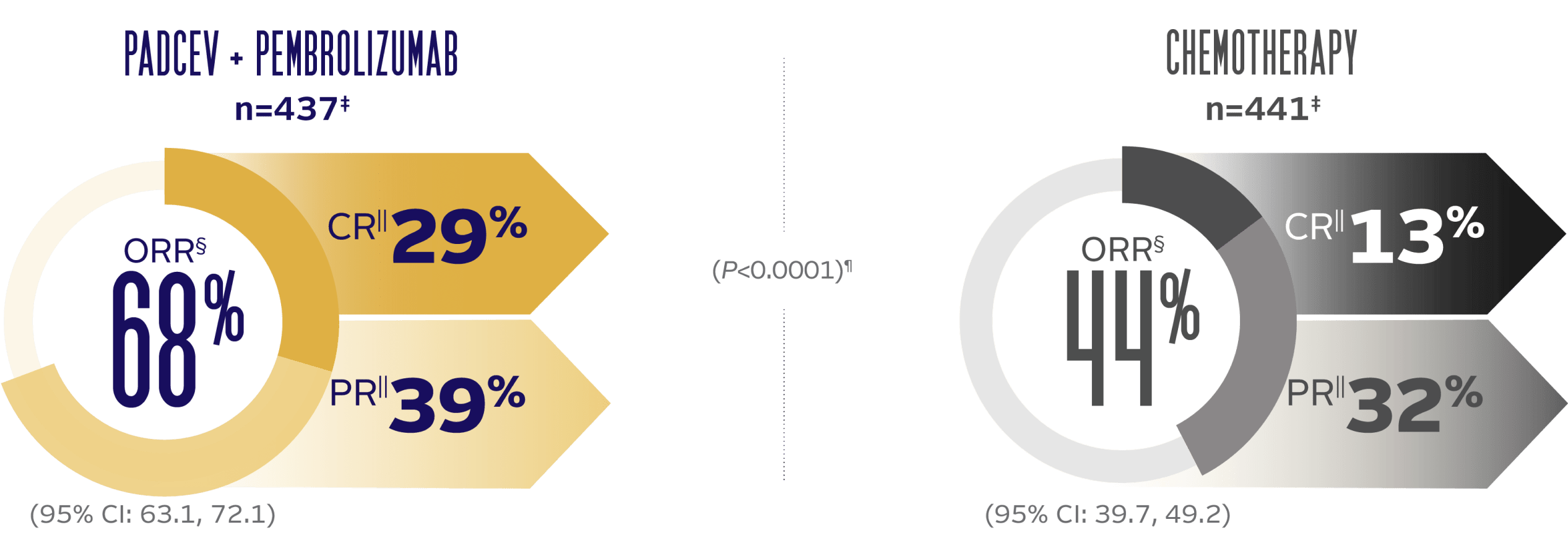
PADCEV + pembrolizumab delivered superior ORR vs chemotherapy1
Nearly 30% of patients achieved a complete response with PADCEV + pembrolizumab
ORR BY BICR PER RECIST v1.1
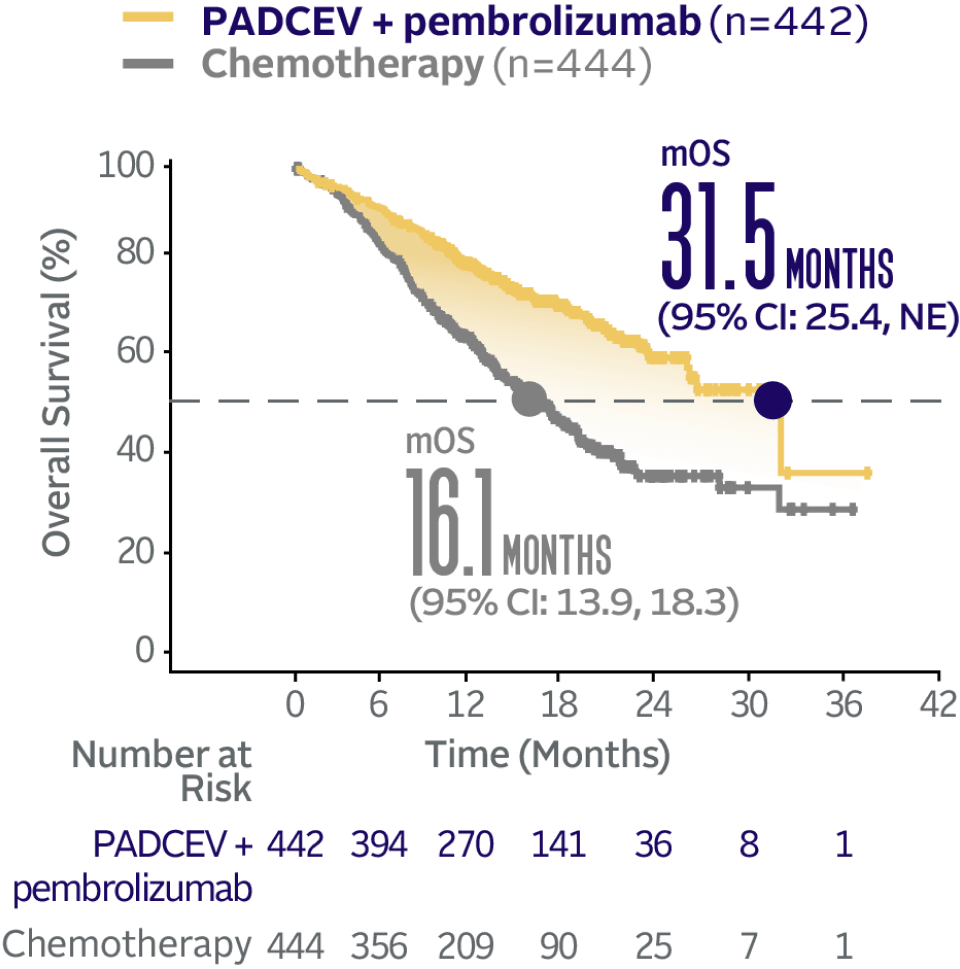
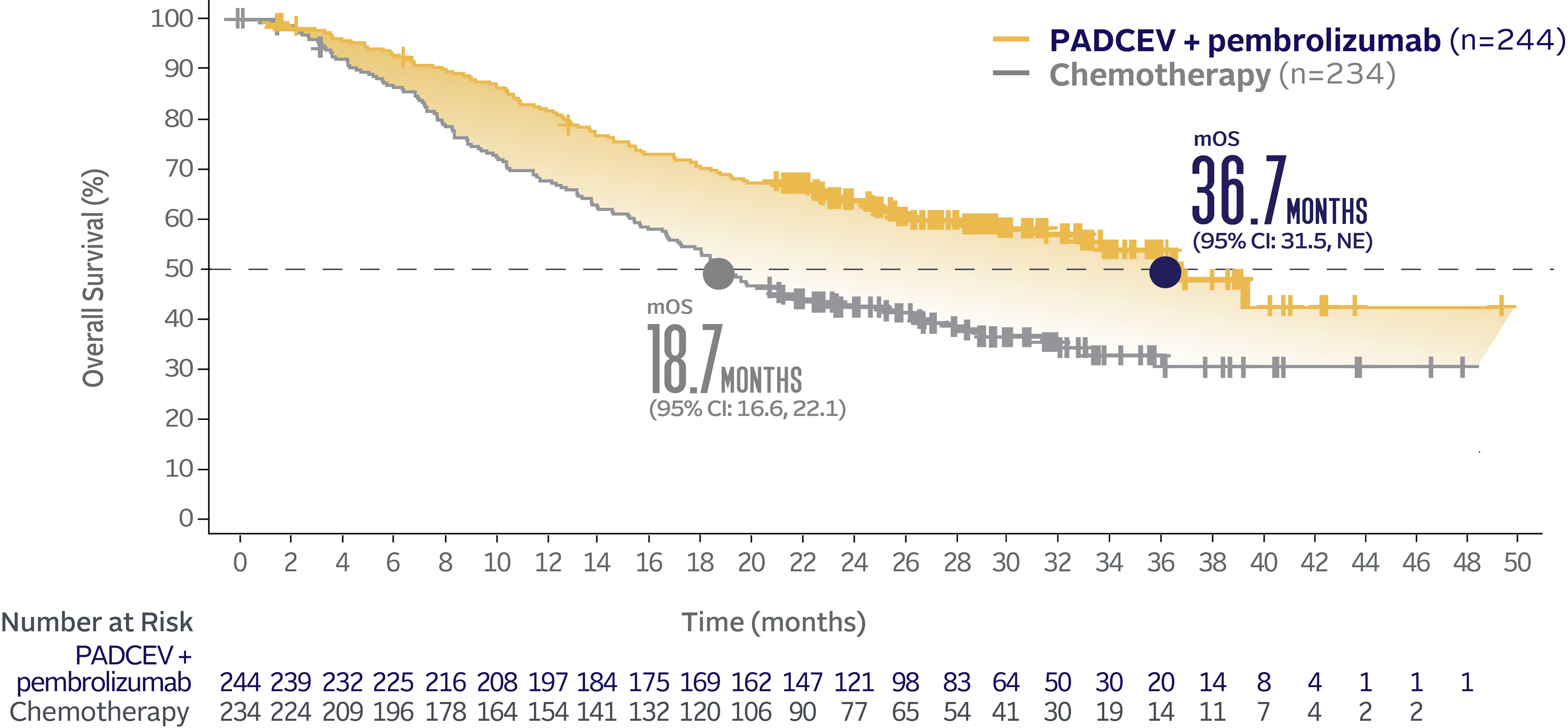
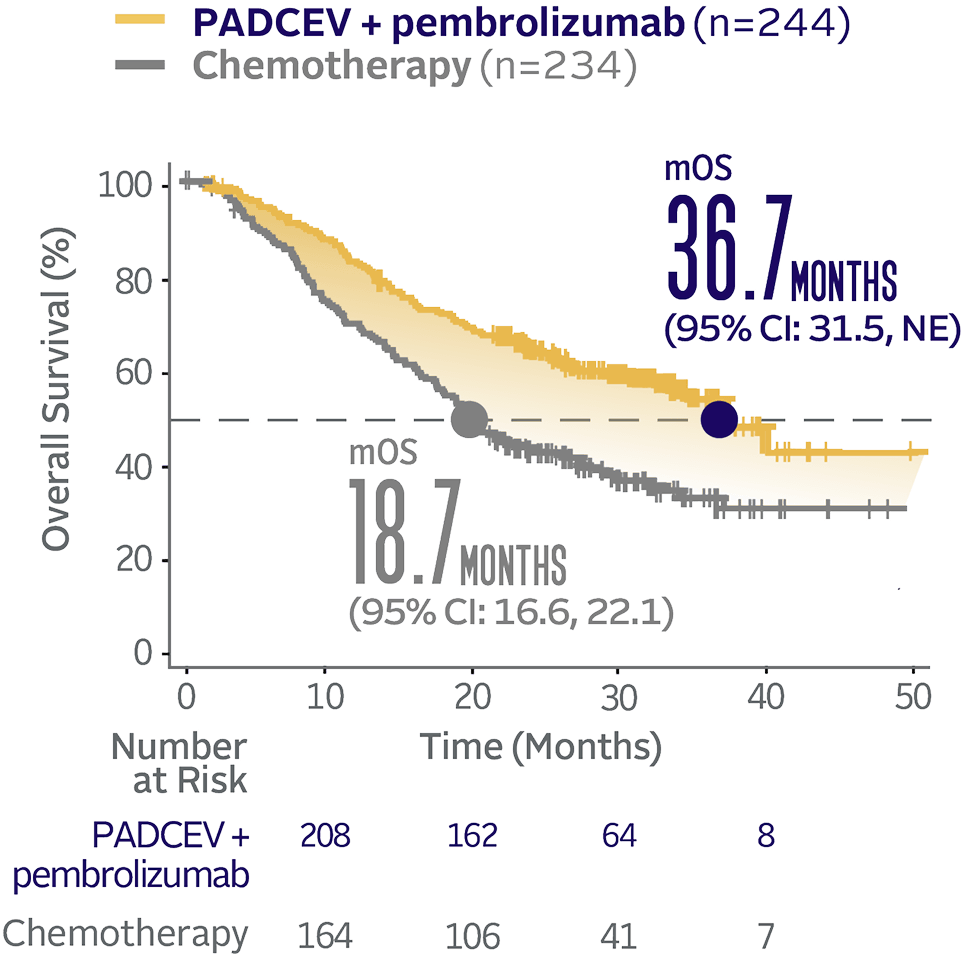
Overall survival data at 29.1 months median follow-up5
The updated analyses with additional ~1 year of follow-up are provided only as descriptive clinical information and no conclusions can be drawn.5
Long-term follow-up OS† (HR=0.51; 95% CI: 0.43, 0.61‡)5
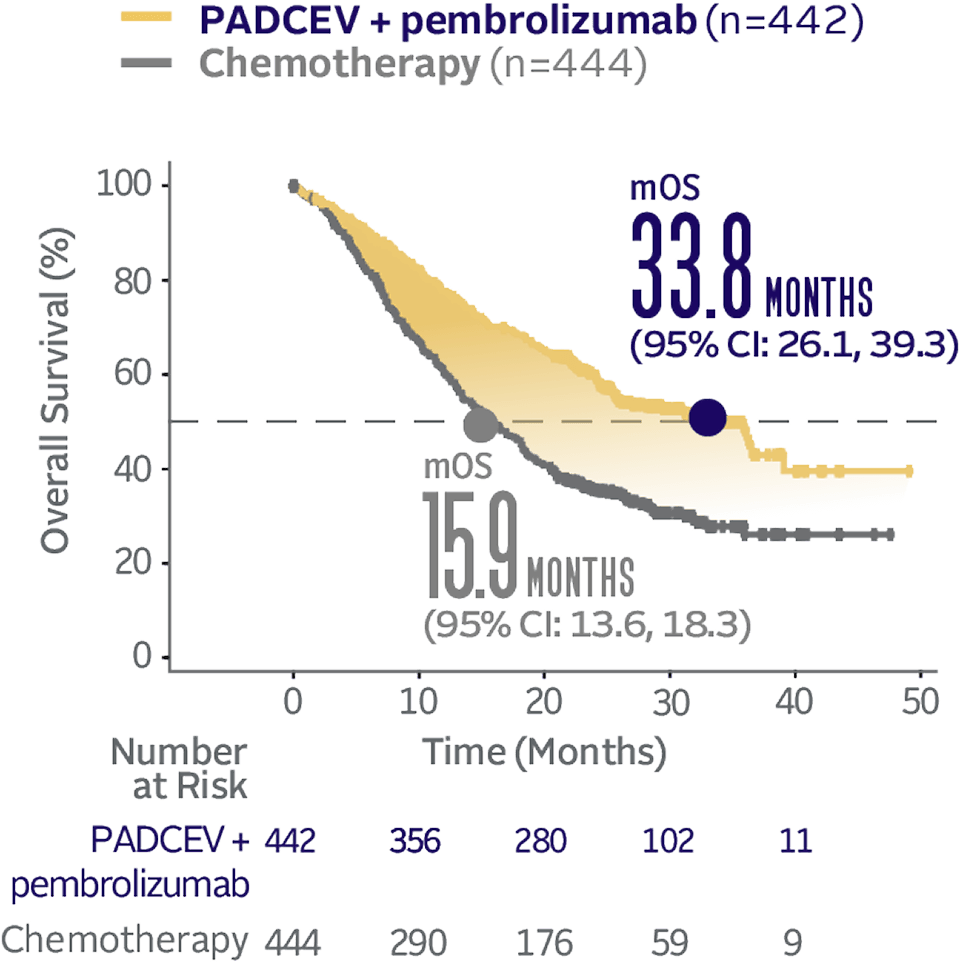
The median follow-up time was 29.1 months (range: 0.1, 49.4).5
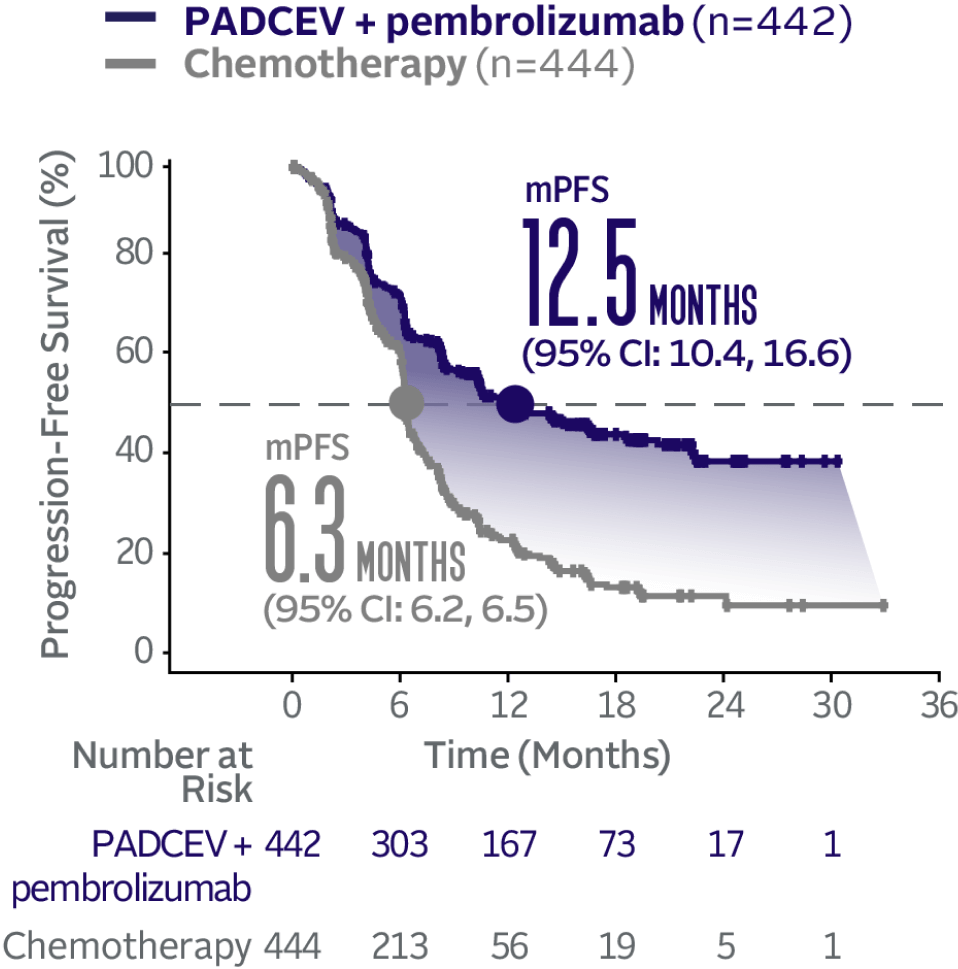
Progression-free survival data at 29.1 months median follow-up5
The updated analyses with additional ~1 year of follow-up are provided only as descriptive clinical information and no conclusions can be drawn.
Long-term follow-up PFS† (HR=0.48; 95% CI: 0.41, 0.57‡)5
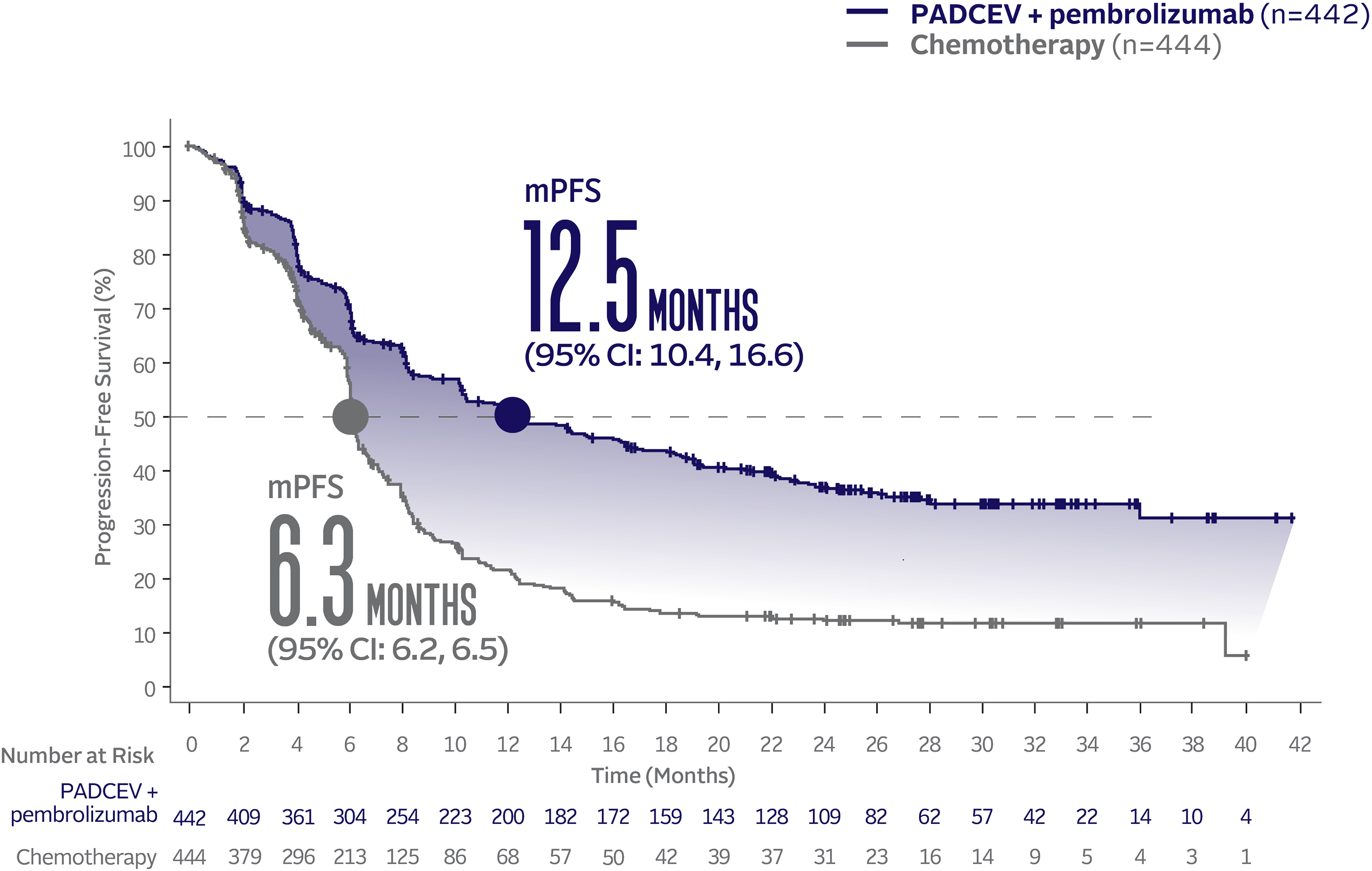
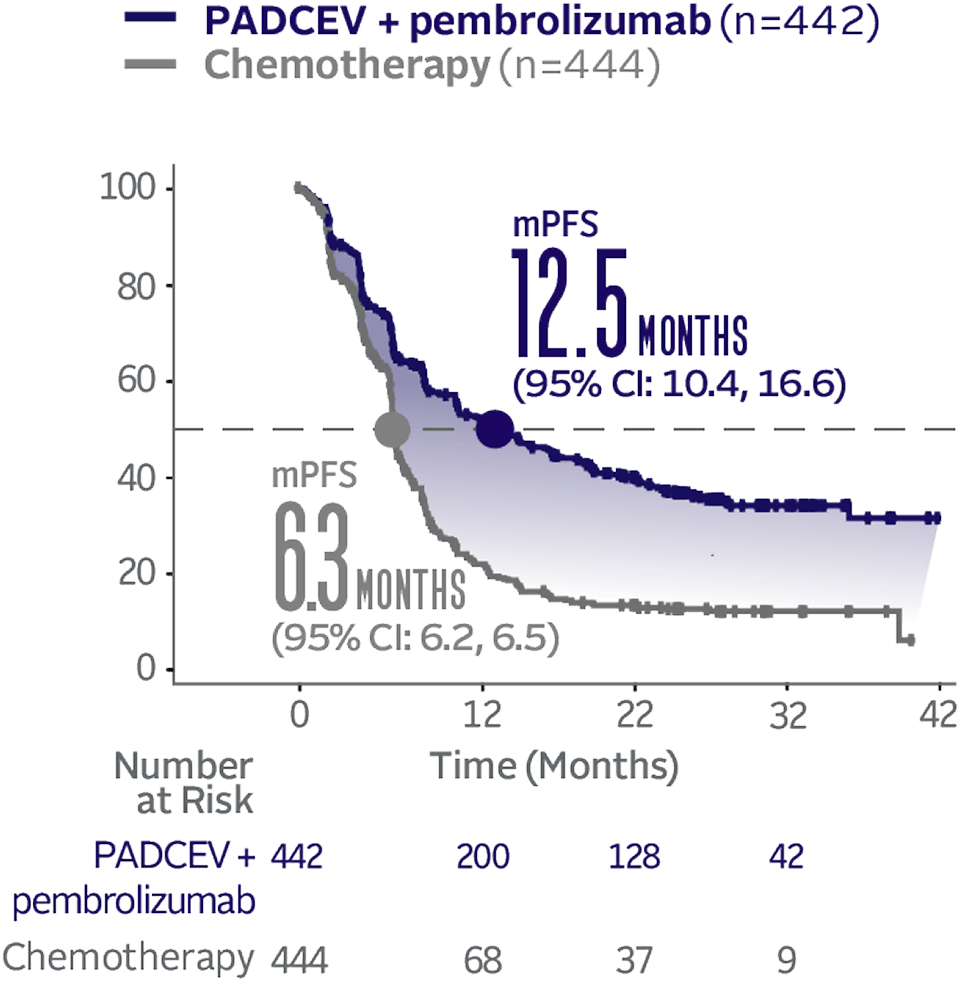
The median follow-up time was 29.1 months (range: 0.1, 49.4).5
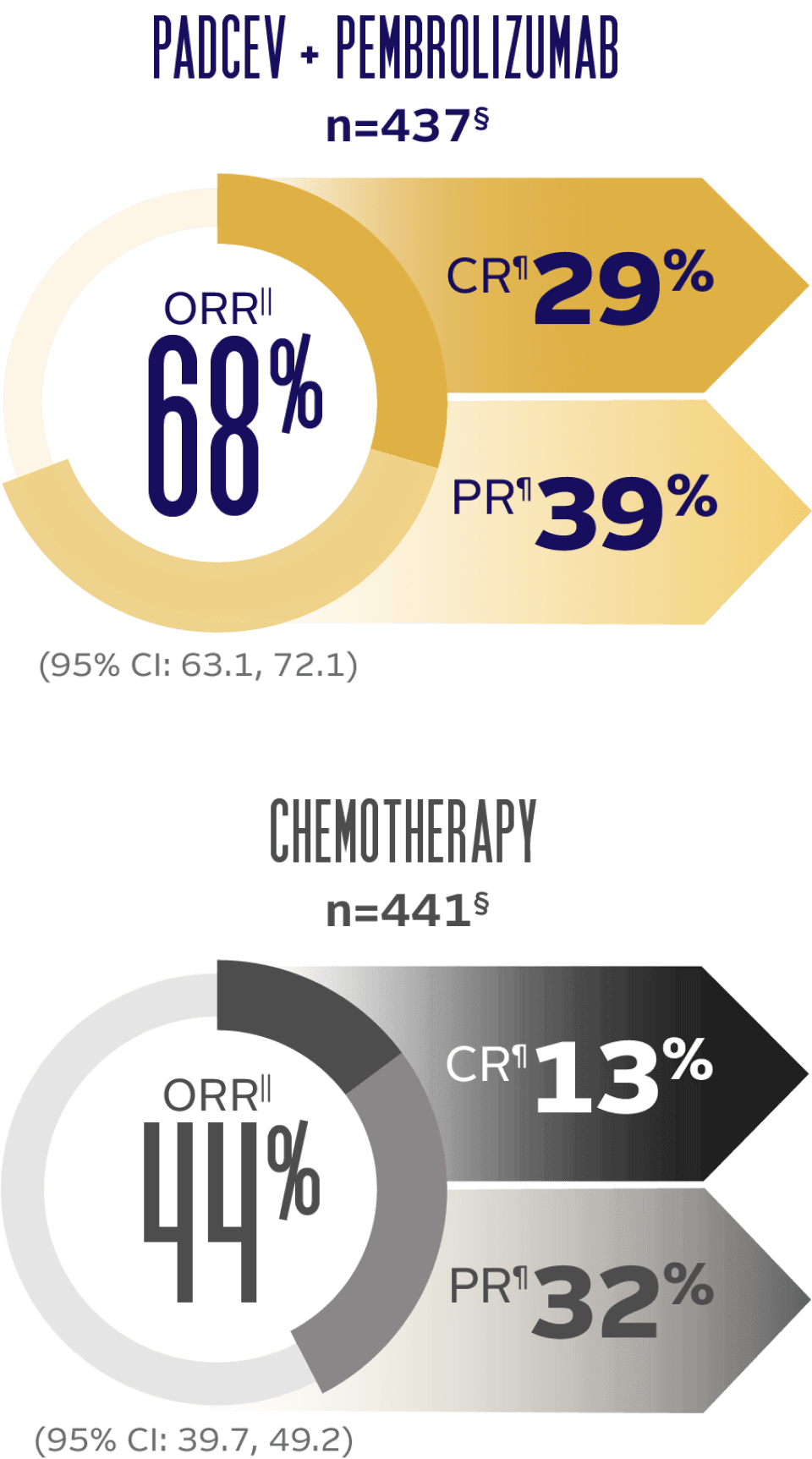
Objective response rate and duration of response (DOR) at 29.1 months median follow-up5
The updated analyses with additional ~1 year of follow-up are provided only as descriptive clinical information and no conclusions can be drawn.
LONG-TERM FOLLOW-UP: ORR BY BICR PER RECIST v1.1
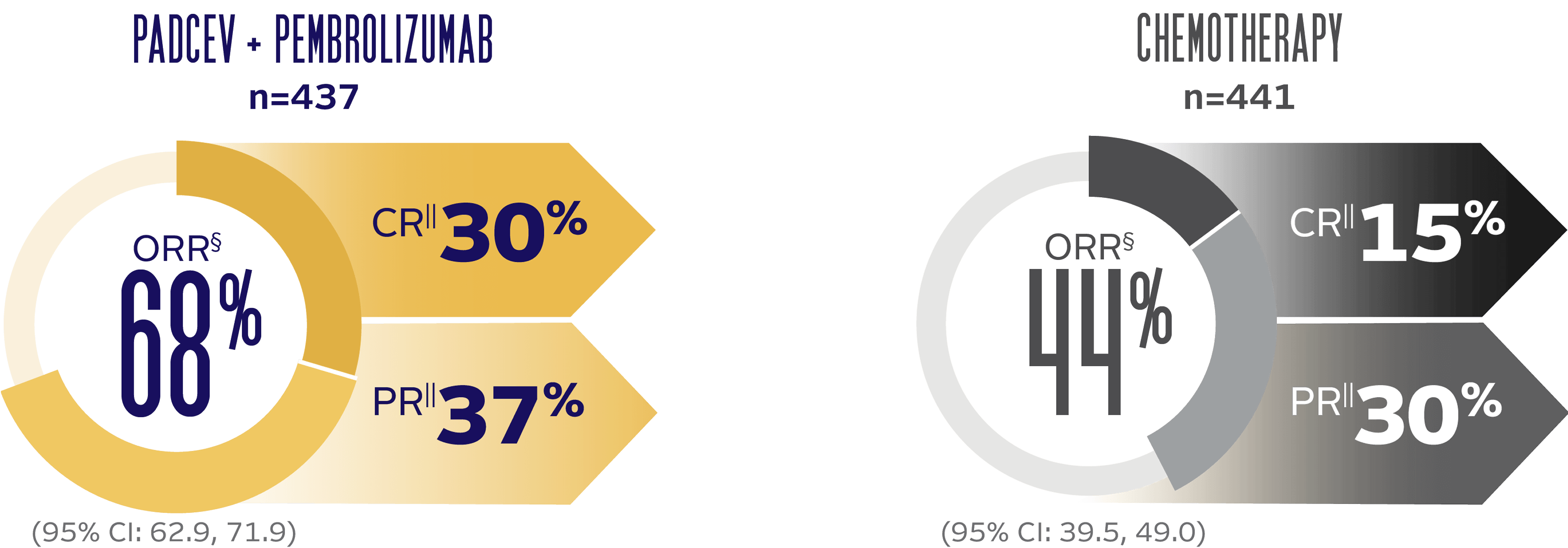
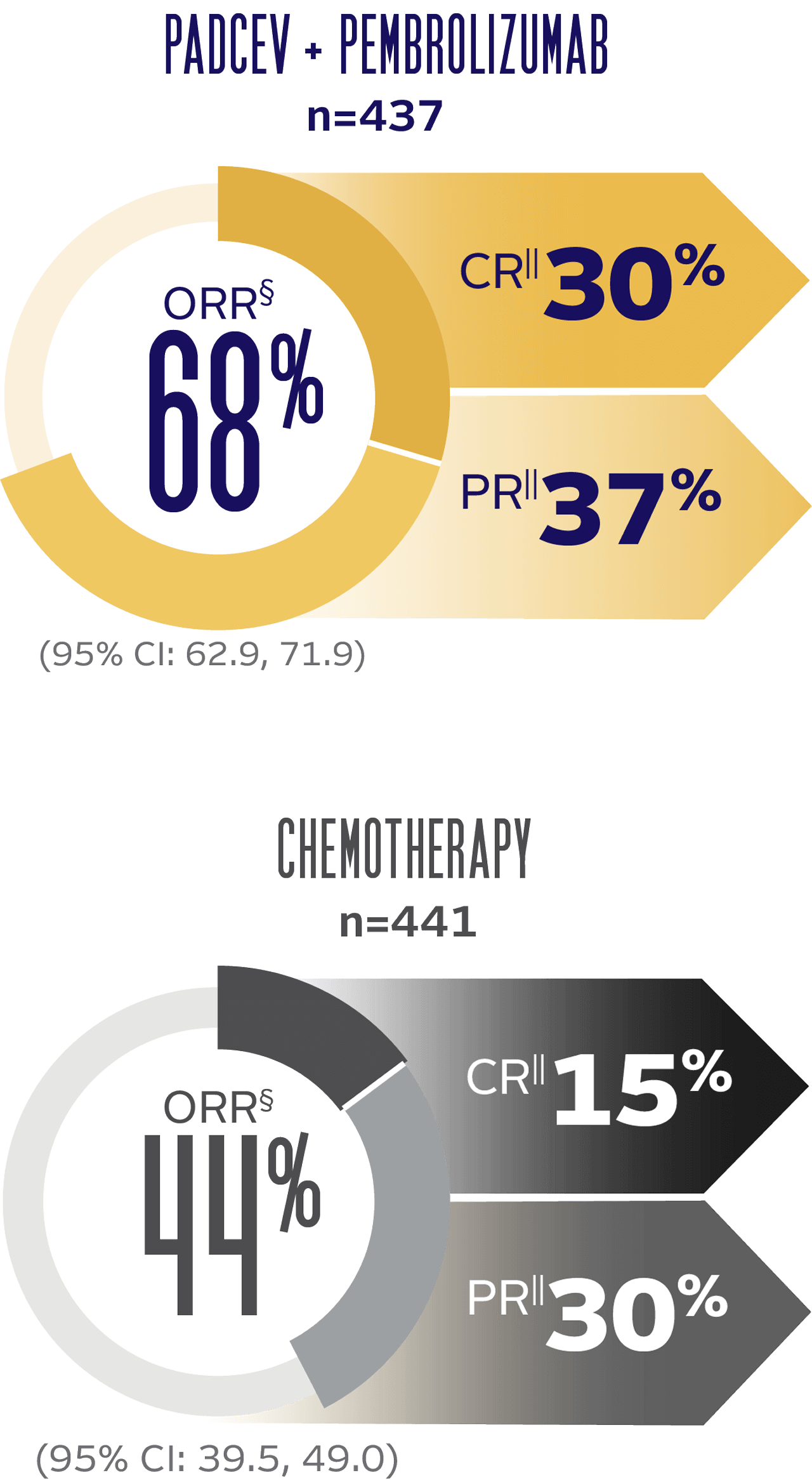
LONG-TERM FOLLOW-UP: MEDIAN DOR (for patients with a CR or PR)†
DOR was evaluated for responders only; therefore, randomization is no longer preserved. The analysis is descriptive only and no comparison between arms can be made.


The median follow-up time was 29.1 months (range 0.1, 49.4).5
Safety data were generally consistent between the initial analysis and the 29.1 month follow-up long-term analysis5

SUBGROUP DATA
Efficacy results were consistent across all stratified patient subgroups1,2,8
Prespecified stratification factors for randomization included:
- Cisplatin eligibility/ineligibility
- Presence/absence of liver metastases
- High (CPS ≥10)/low (CPS <10) PD‑L1 expression
Subgroup analyses were exploratory in nature. This study was not powered to detect differences between treatments based on prespecified subgroups. Results of the exploratory subgroup analyses are descriptive but not conclusive, are not controlled for type I error, and should be interpreted with caution.
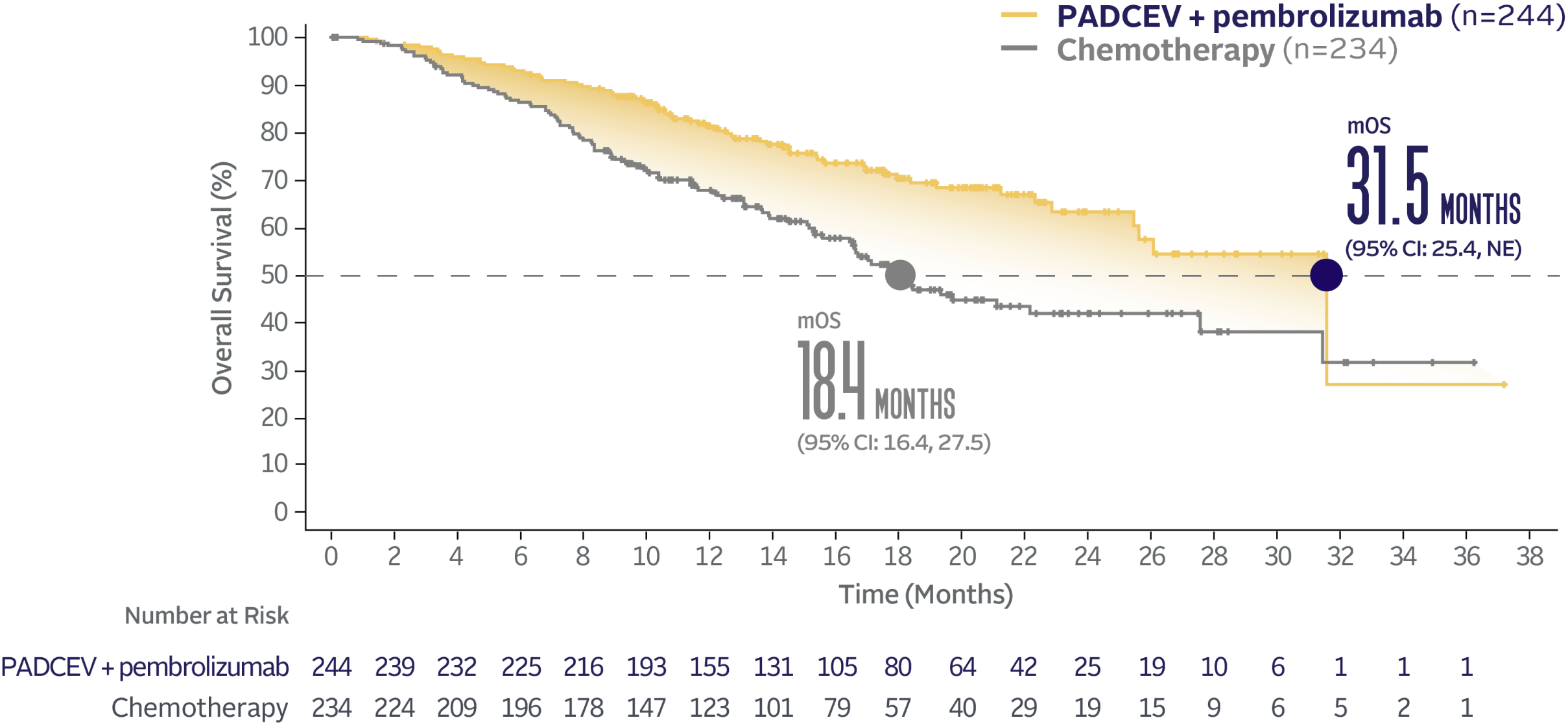
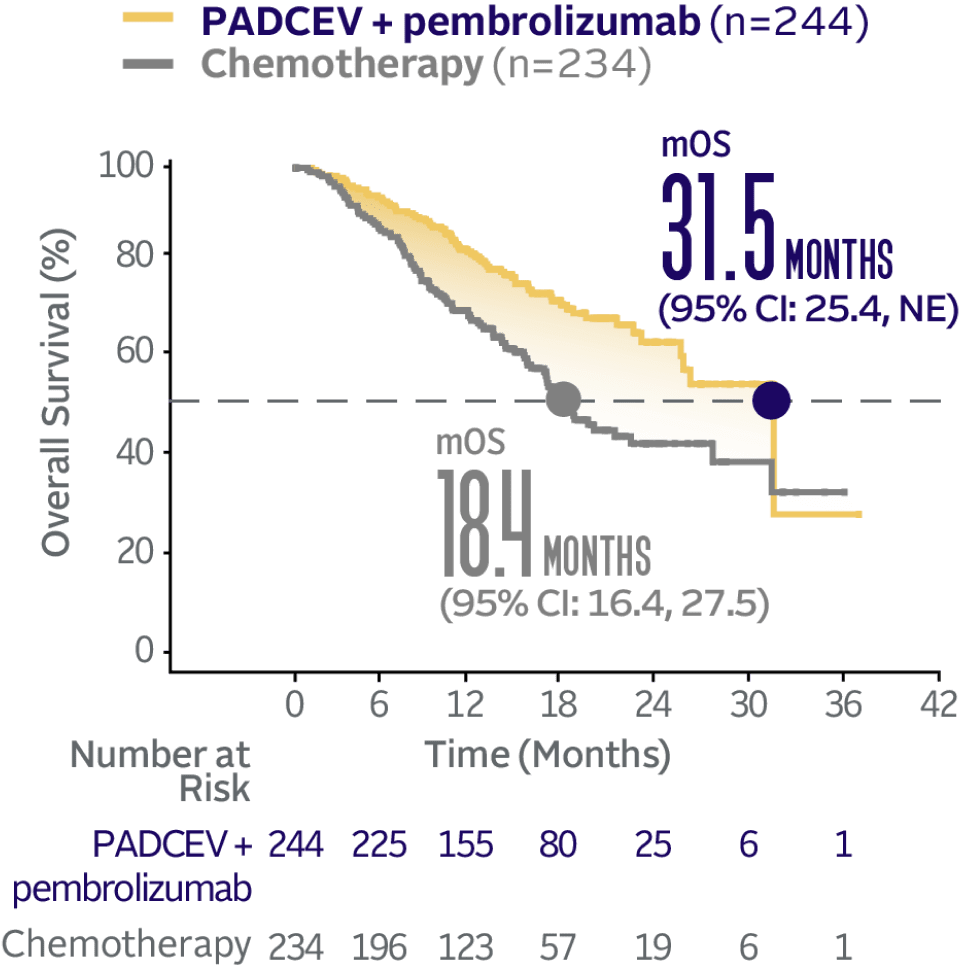
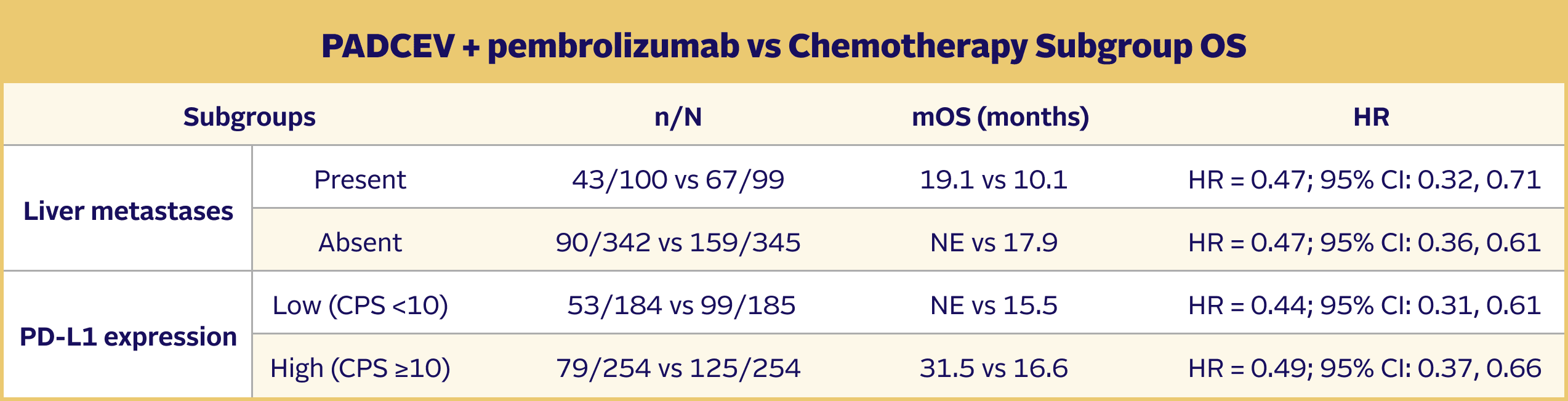
EV‑302 Subgroup OS:
CISPLATIN ELIGIBLE
(HR=0.53; 95% CI: 0.39, 0.72)
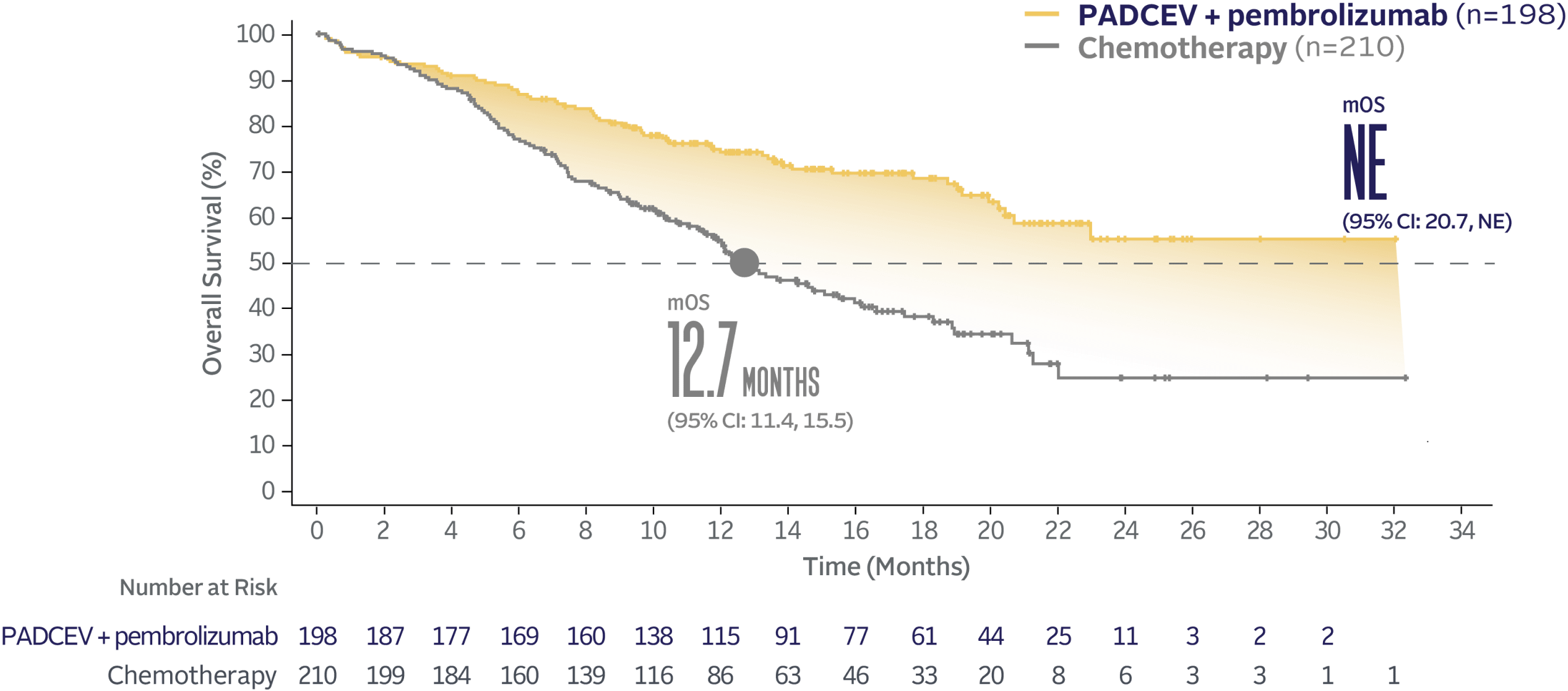
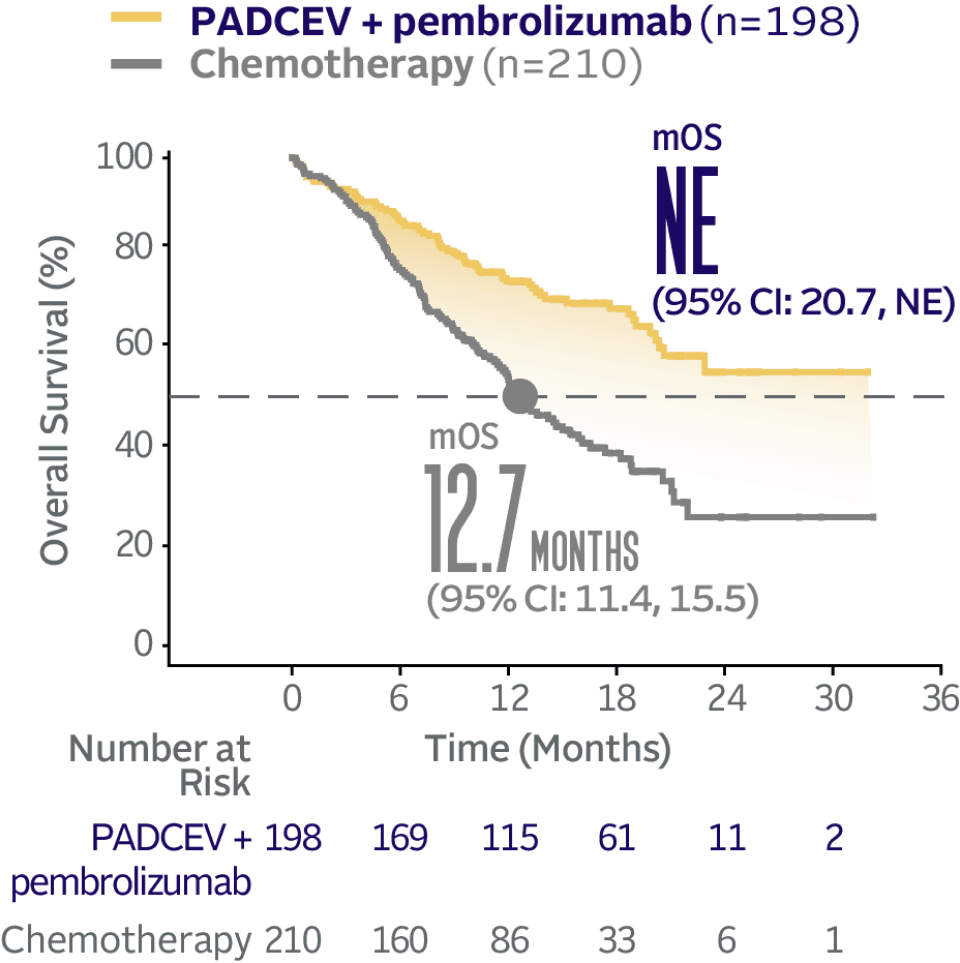
EV‑302 Subgroup OS:
CISPLATIN INELIGIBLE
(HR=0.43; 95% CI: 0.31, 0.59)
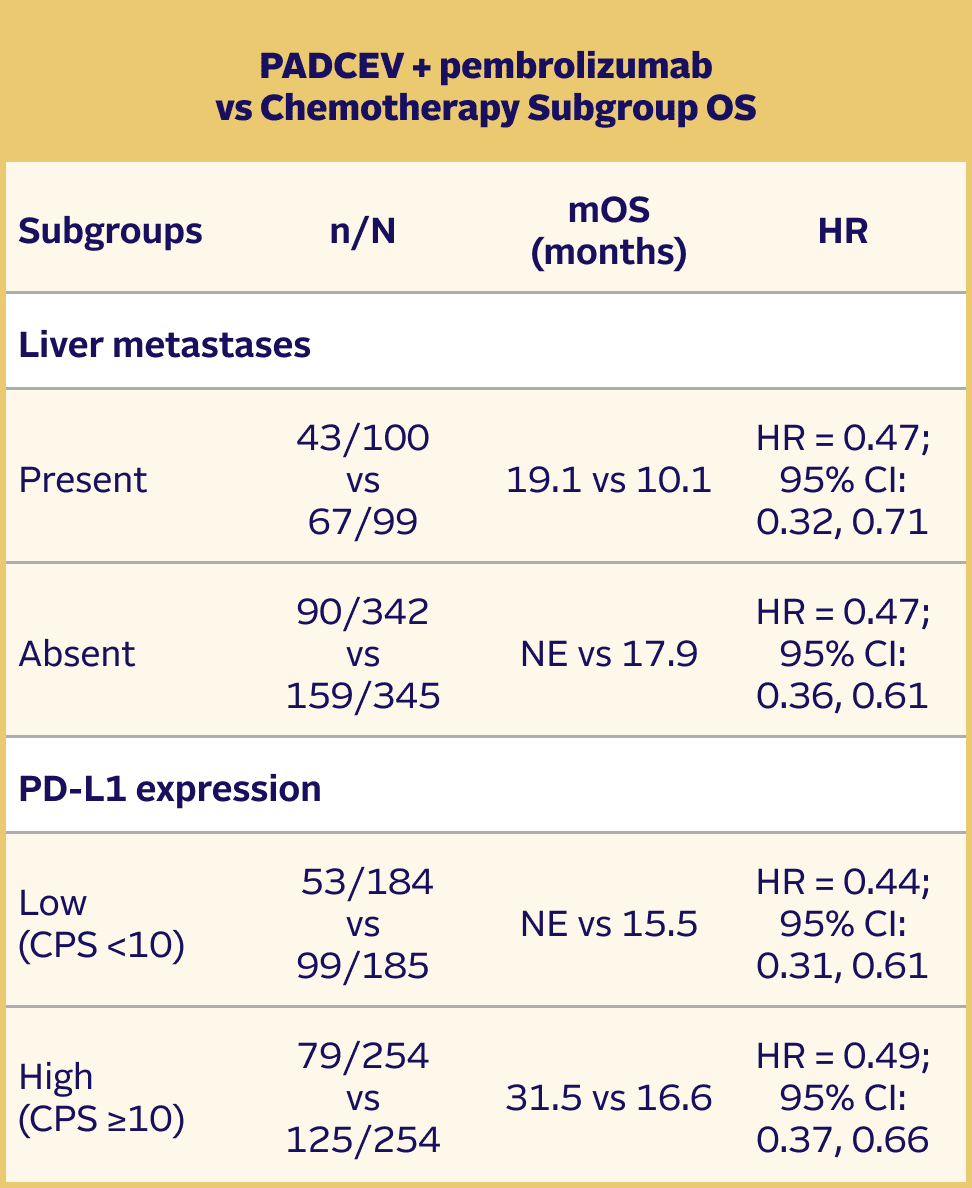
Long-term follow-up: OS in prespecified subgroups5,9
The updated analyses with additional ~1 year of follow-up are provided only as descriptive clinical information and no conclusions can be drawn.
Subgroup analyses were exploratory in nature. This study was not powered to detect differences between treatments based on prespecified subgroups. Results of the exploratory subgroup analyses are descriptive but not conclusive, are not controlled for type I error, and should be interpreted with caution.
EV-302 Long-Term Follow-Up Subgroup OS: CISPLATIN ELIGIBLE (HR=0.54; 95% CI: 0.42, 0.70)
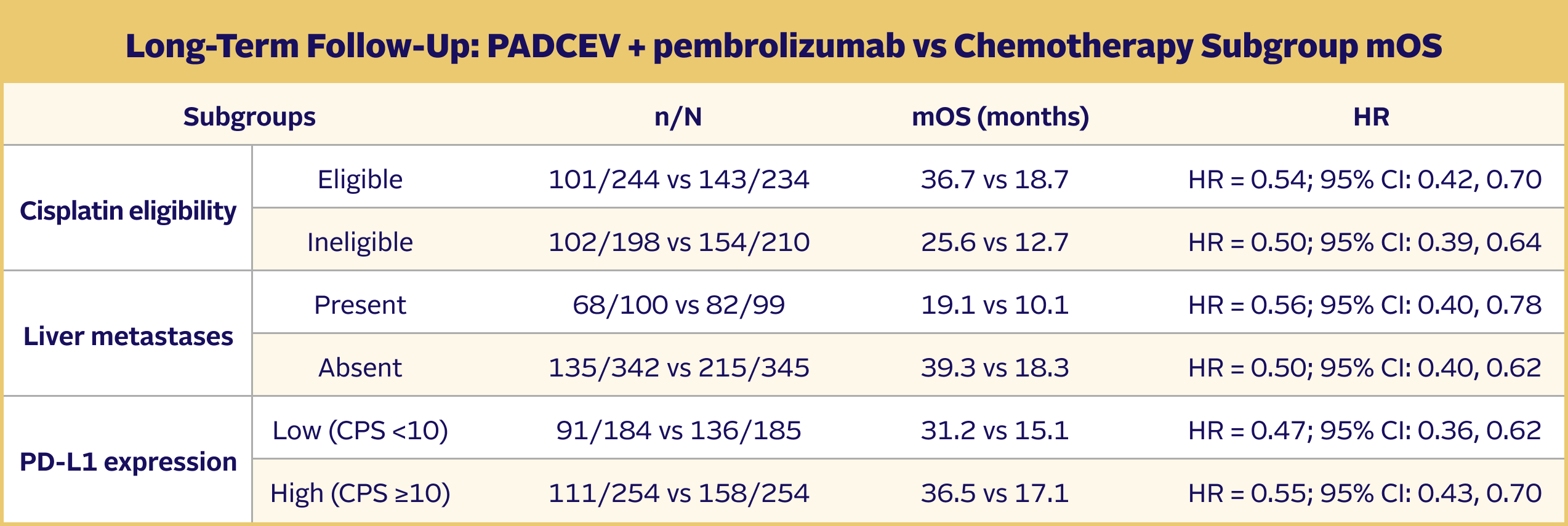
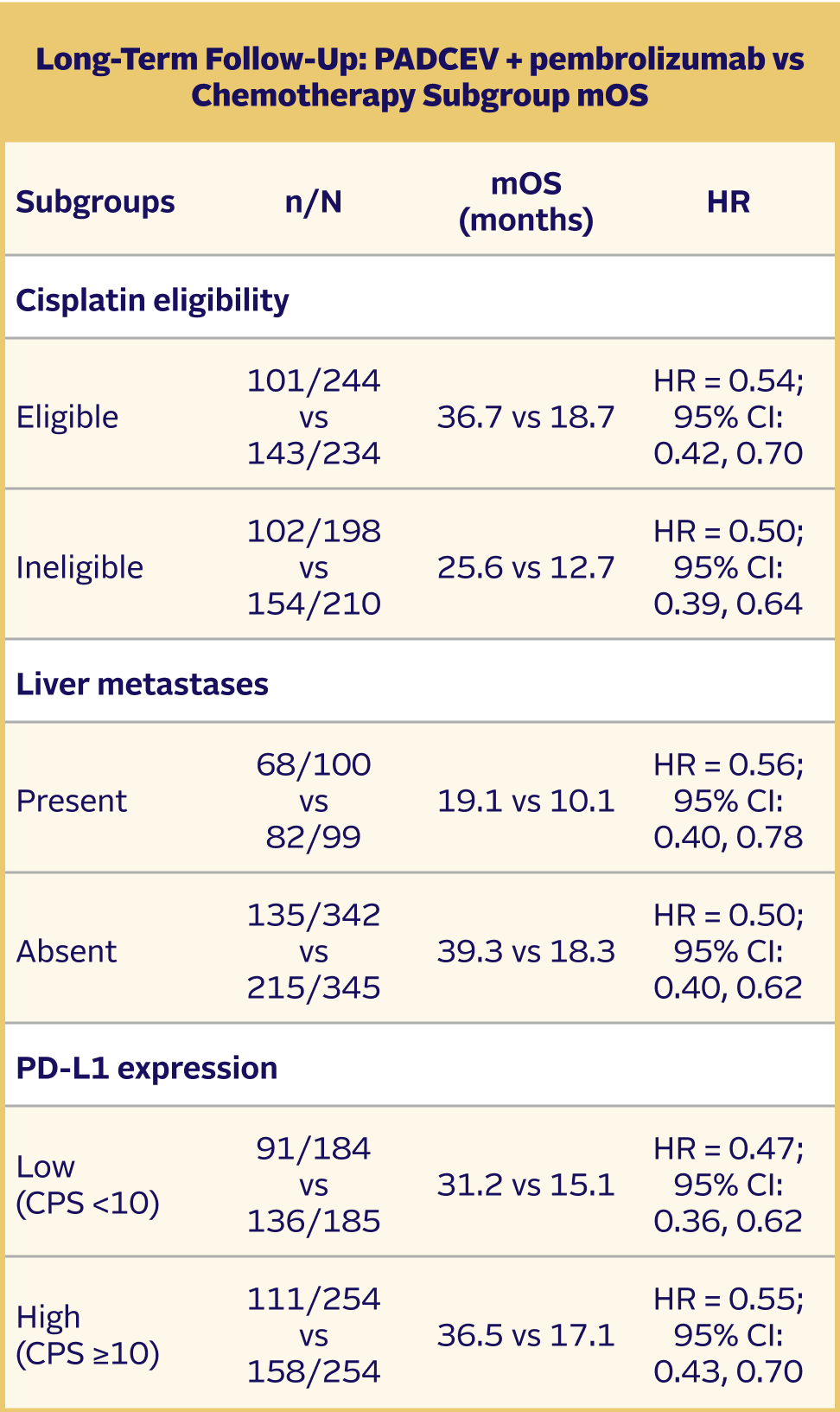
The median follow-up time was 29.1 months (range: 0.1, 49.4).5
TRIAL DESIGN
EV‑302: A pivotal phase 3 trial in 1L la/mUC1,2,8,10
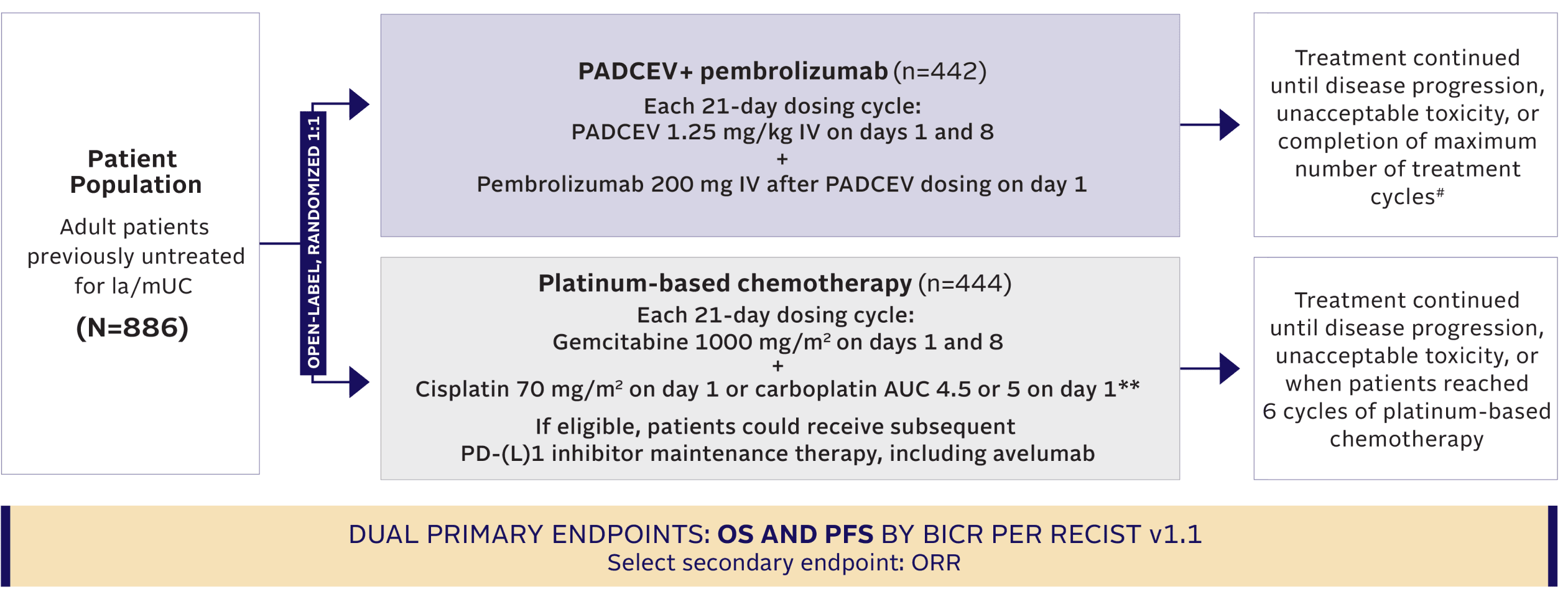
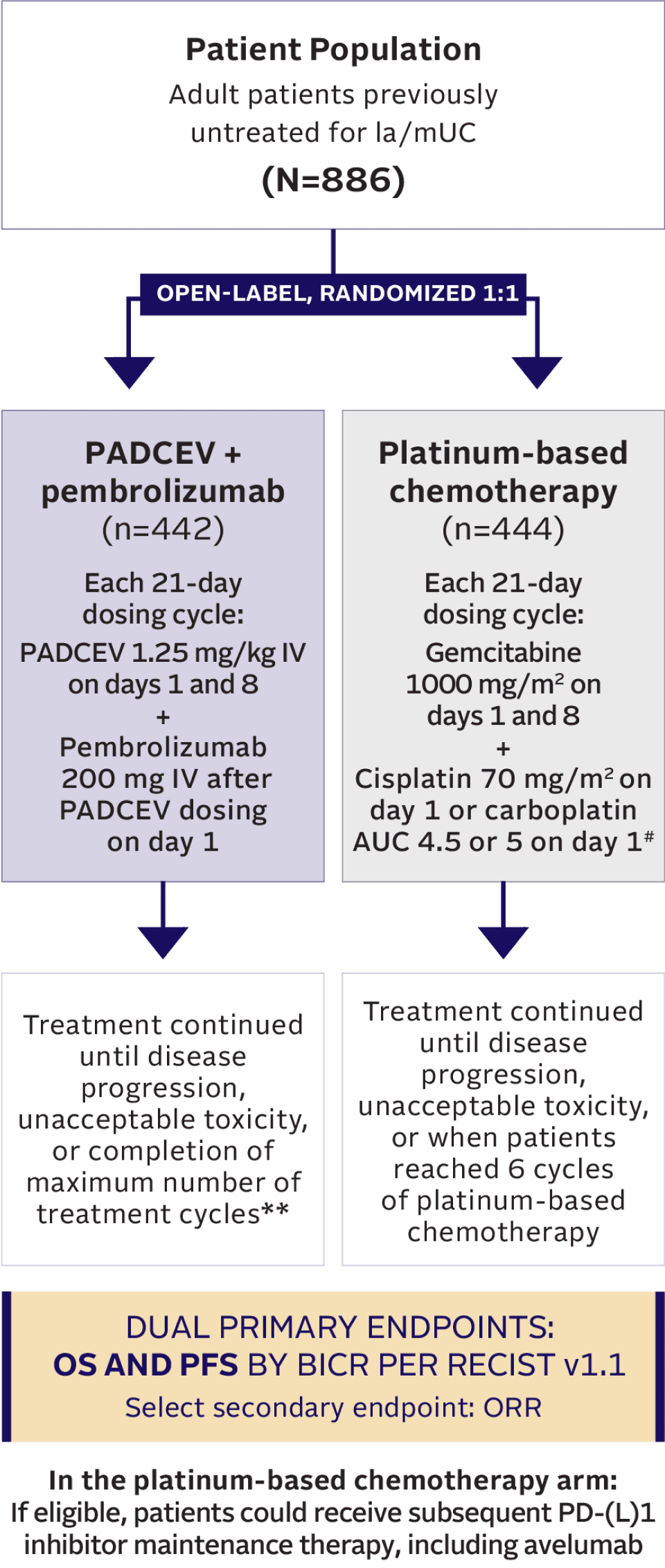
Maintenance therapy (eg, avelumab) was permitted following completion and/or discontinuation of platinum-based chemotherapy, if locally available, and provided the patient was deemed appropriate by the investigator.2,10
Maintenance therapy (eg, avelumab) was permitted following completion and/or discontinuation of platinum-based chemotherapy, if locally available, and provided the patient was deemed appropriate by the investigator.2,10
Key inclusion criteria10
- Must be eligible to receive either cisplatin or carboplatin††
- ECOG PS 0, 1, 2‡
Prespecified stratification factors for randomization included1,2:
- Cisplatin eligibility/ineligibility
- Presence/absence of liver metastases
- High (CPS ≥10)/low (CPS <10) PD‑L1 expression
Key exclusion criteria1,2
- Prior PD‑(L)1 inhibitor therapy
- Active CNS metastases
- Ongoing sensory or motor neuropathy Grade ≥2
- Uncontrolled diabetes defined as HbA1c ≥8% or HbA1c ≥7% with associated diabetes symptoms
- Prior autoimmune disease requiring systemic treatment within the past 2 years
Prespecified stratification factors for randomization included1,2:
- Cisplatin eligibility/ineligibility
- Presence/absence of liver metastases
- High (CPS ≥10)/low (CPS <10) PD‑L1 expression
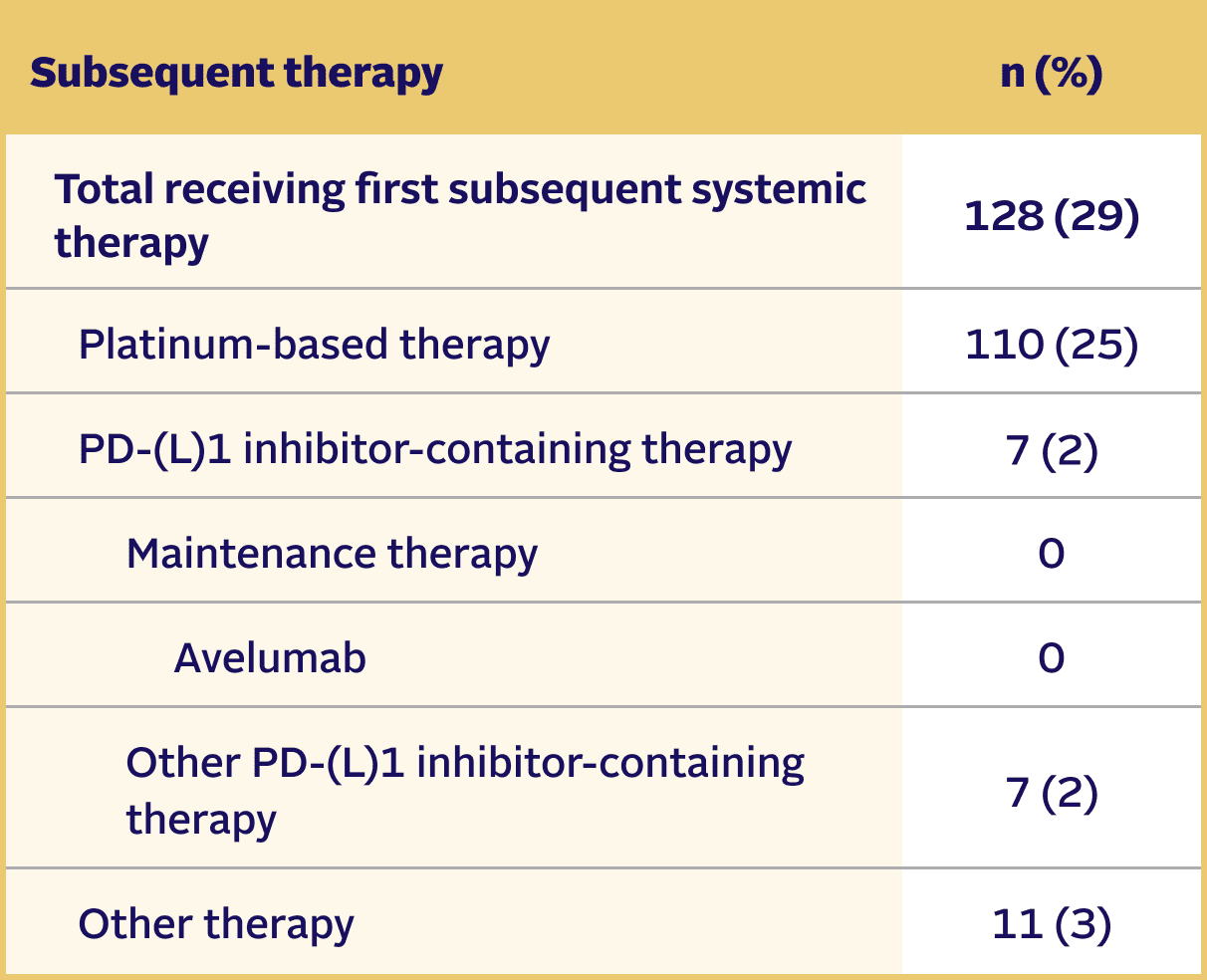
SUBSEQUENT THERAPY
Subsequent therapies in the EV‑302 trial5,8
At the time of data cutoff,
29% of patients in the PADCEV + pembrolizumab arm (n=442) received subsequent systemic therapy

At the time of data cutoff,
66% of patients in the chemotherapy arm (n=444) received subsequent systemic therapy
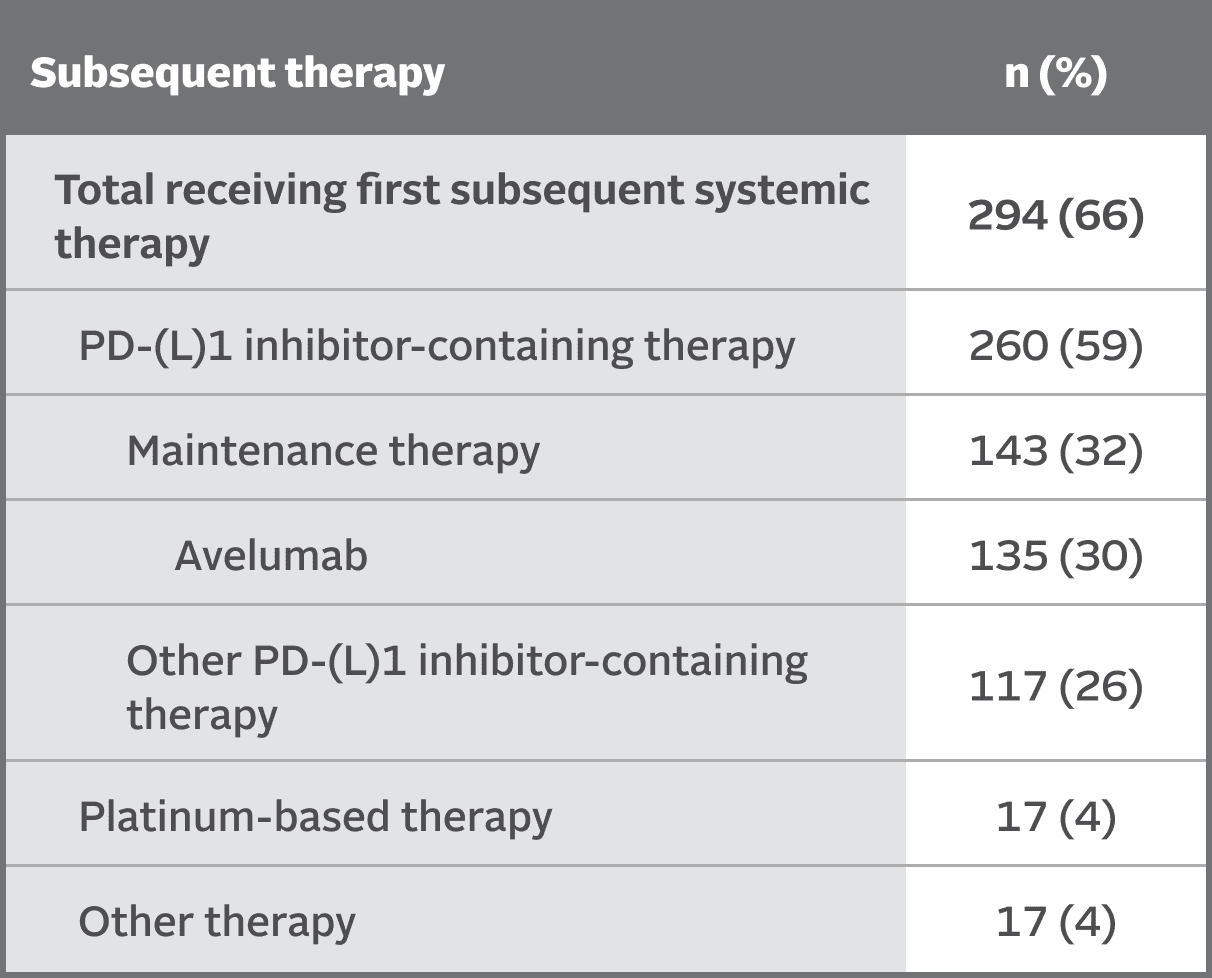
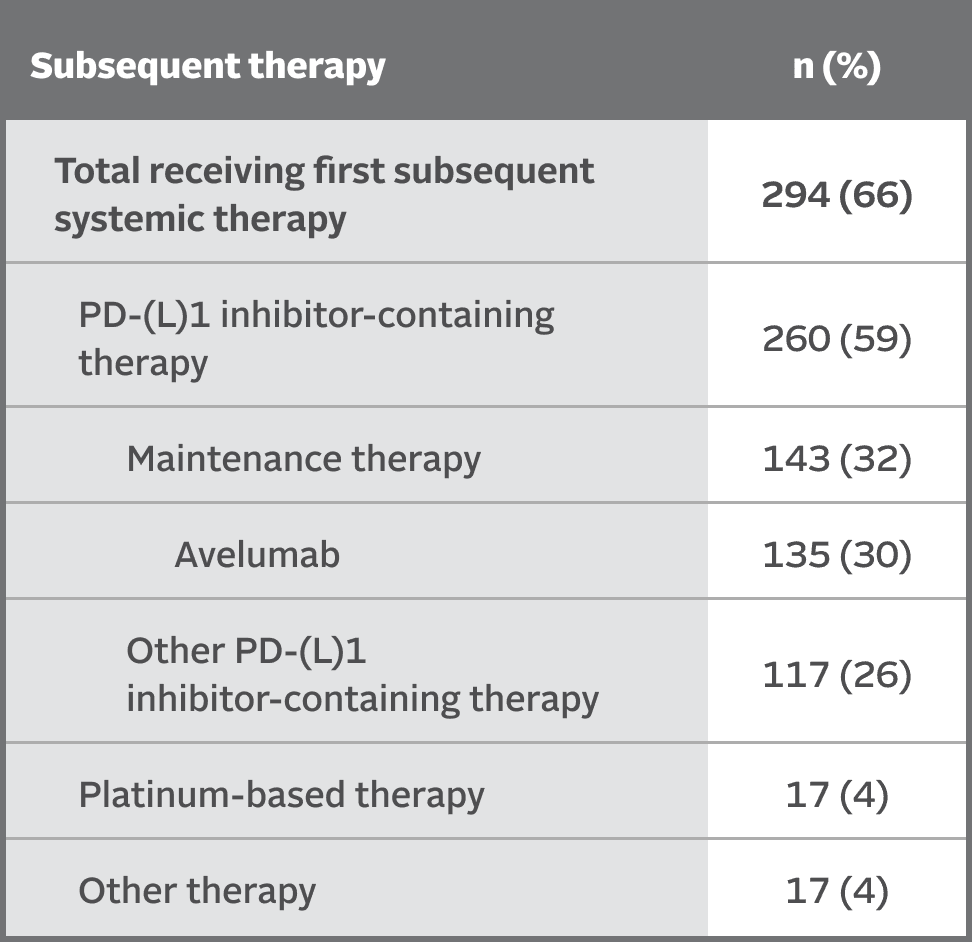
Maintenance therapy (eg, avelumab) was permitted following completion and/or discontinuation of platinum-based chemotherapy, if locally available, and provided the patient was deemed appropriate by the investigator.2,10
PATIENT CHARACTERISTICS
Treatment arms were balanced and representative of the la/mUC patient population2
Select Baseline
Patient
Characteristics
PADCEV +
pembrolizumab
(n=442)
Chemotherapy
(n=444)
Median age, years
(range)
69 (37, 87)
69 (22, 91)
Sex, n (%)
Male
344 (78)
336 (76)
Female
98 (22)
108 (24)
Race or ethnic group,* n (%)
Asian
99 (22)
92 (21)
Black or African American
3 (1)
7 (2)
White
308 (70)
290 (65)
Other†
5 (1)
8 (2)
Unknown or not reported
27 (6)
47 (11)
Geographic region, n (%)
North America
103 (23)
85 (19)
Europe
172 (39)
197 (44)
Rest of world
167 (38)
162 (37)
ECOG PS,‡ n (%)
0
223 (51)
215 (48)
1
204 (46)
216 (49)
2
15 (3)
11 (3)
Data missing
0
2 (1)
Body-mass index,§ n (%)
<25
206 (47)
185 (42)
25 to <30
144 (33)
155 (35)
≥30
89 (20)
101 (23)
Data missing
3 (1)
3 (1)
Creatinine clearance,|| n (%)
≥60 mL/min
249 (56)
257 (58)
<60 mL/min
193 (44)
187 (42)
Number of Bajorin risk factors,¶ n (%)
0
179 (41)
183 (41)
1
263 (60)
259 (58)
Data missing
0
2 (1)
H score of Nectin-4 expression#
Number of patients tested
394
406
Median score (range)
280 (0, 300)
270 (0, 300)
Disease status at randomization, n (%)
Locally advanced
21 (5)
24 (5)
Metastatic
421 (95)
420 (95)
Primary site of origin of disease, n (%)
Upper tract
135 (31)
104 (23)
Lower tract
305 (69)
339 (76)
Unknown
2 (1)
1 (0)
Histologic type, n (%)
Urothelial carcinoma
379 (86)
373 (84)
Urothelial carcinoma, mixed types**
50 (11)
53 (12)
Variant urothelial carcinoma only
4 (1)
7 (2)
Unknown
9 (2)
11 (3)
Sites of metastasis, n (%)
Lymph node only
103 (23)
104 (23)
Visceral site
318 (72)
318 (72)
Bone
81 (18)
102 (23)
Liver
100 (23)
99 (22)
Lung
170 (39)
157 (35)
Cisplatin eligibility status,†† n (%)
Cisplatin eligible
240 (54)
242 (55)
Cisplatin ineligible
202 (46)
202 (46)
PD‑L1 expression,‡‡ n/n (%)
High (CPS ≥10)
254/438 (58)
254/439 (58)
Low (CPS <10)
184/438 (42)
185/439 (42)
1L=first-line; AUC=area under the curve; BICR=blinded independent central review; CI=confidence interval; CNS=central nervous system; CPS=Combined Positive Score; CR=complete response; ECOG=Eastern Cooperative Oncology Group; GFR=glomerular filtration rate; HbA1c=hemoglobin A1c; HR=hazard ratio; IHC=immunohistochemistry; IV=intravenous; la/mUC=locally advanced or metastatic urothelial cancer; mOS=median overall survival; mPFS=median progression-free survival; NCI CTCAE=National Cancer Institute Common Terminology Criteria for Adverse Events; NE=not estimable; NYHA=New York Heart Association; ORR=objective response rate; OS=overall survival; PD‑(L)1=programmed death receptor-1 or programmed death-ligand 1; PFS=progression-free survival; PR=partial response; PS=performance status; RECIST=Response Evaluation Criteria in Solid Tumors; WHO=World Health Organization.
References: 1. PADCEV [package insert]. Northbrook, IL: Astellas Pharma US, Inc. 2. Powles T, Valderrama BP, Gupta S, et al; for the EV‑302 Trial Investigators. Enfortumab vedotin and pembrolizumab in untreated advanced urothelial cancer. N Engl J Med 2024;390(10):875-88. 3. Powles T, Bellmunt J, Comperat E, et al; for the ESMO Guidelines Committee. ESMO Clinical Practice Guideline interim update on first-line therapy in advanced urothelial carcinoma. Ann Oncol 2024;35(6):485-90. 4. Feldman AS, Lee RJ, Miyamoto DT, Dahl DM, Efstathiou JA. Cancer of the bladder, ureter, and renal pelvis. In: DeVita Jr VT, Lawrence TS, Rosenberg SA, eds. DeVita, Hellman, and Rosenberg's Cancer: Principles and Practice of Oncology. 12th ed. Wolters Kluwer Health; 2023:756-83. 5. Pfizer Inc. and Astellas. PADCEV. Data on File. 6. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Bladder Cancer V.1.2025. © National Comprehensive Cancer Network, Inc. 2025. All rights reserved. Accessed March 25, 2025. To view the most recent and complete version of the guideline, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way. 7. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45(2):228-47. 8. Supplement to: Powles T, Valderrama BP, Gupta S, et al; for the EV‑302 Trial Investigators. Enfortumab vedotin and pembrolizumab in untreated advanced urothelial cancer. N Engl J Med 2024;390(10):875-88. 9. Powles TB, Van der Heijden MS, Loriot Y, et al. EV-302: Updated analysis from the phase 3 global study of enfortumab vedotin in combination with pembrolizumab (EV+P) vs chemotherapy (chemo) in previously untreated locally advanced or metastatic urothelial carcinoma (la/mUC). Presented at: American Society of Clinical Oncology Genitourinary Cancers Symposium; February 13-15, 2025; San Francisco, CA. 10. Protocol for: Powles T, Valderrama BP, Gupta S, et al; for the EV‑302 Trial Investigators. Enfortumab vedotin and pembrolizumab in untreated advanced urothelial cancer. N Engl J Med 2024;390(10):875-88.
Important Safety Information/Indications
INDICATIONS
PADCEV, in combination with pembrolizumab or pembrolizumab and berahyaluronidase alfa-pmph, as neoadjuvant treatment and then continued after cystectomy as adjuvant treatment, is indicated for the treatment of adult patients with muscle invasive bladder cancer (MIBC) who are ineligible for cisplatin-containing chemotherapy.
PADCEV, in combination with pembrolizumab or pembrolizumab and berahyaluronidase alfa-pmph, is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer (mUC).
PADCEV, as a single agent, is indicated for the treatment of adult patients with locally advanced or mUC who: